Examples
In the following a few code-snippets are shown which should help you getting started with xrayutilities. Not all of the codes shown in the following will be run-able as stand-alone script. For fully running scripts look in the GitHub examples or in the download.
Reading data from data files
The io submodule provides classes for reading x-ray diffraction data in
various formats. In the following few examples are given.
Reading SPEC files
- Working with spec files in xrayutilities can be done in two distinct ways.
parsing the spec file for scan headers; and parsing the data only when needed
parsing the spec file for scan headers; parsing all data and dump them to an HDF5 file; reading the data from the HDF5 file.
Both methods have their pros and cons. For example when you parse the spec-files over a network connection you need to re-read the data again over the network if using method 1) whereas you can dump them to a local file with method 2). But you will parse data of the complete file while dumping it to the HDF5 file.
Both methods work incremental, so they do not start at the beginning of the file when you reread it, but start from the last position they were reading and work with files including data from linear detectors.
An working example for both methods is given in the following.
1import xrayutilities as xu
2import os
3
4# open spec file or use open SPECfile instance
5try: s
6except NameError:
7 s = xu.io.SPECFile("sample_name.spec", path="./specdir")
8
9# method (1)
10s.scan10.ReadData()
11scan10data = s.scan10.data
12
13# method (2)
14h5file = os.path.join("h5dir", "h5file.h5")
15s.Save2HDF5(h5file) # save content of SPEC file to HDF5 file
16# read data from HDF5 file
17[angle1, angle2], scan10data = xu.io.geth5_scan(h5file, [10],
18 "motorname1",
19 "motorname2")
See also
the fully working example hello world
In the following it is shown how to re-parsing the SPEC file for new scans and reread the scans (1) or update the HDF5 file(2)
1s.Update() # reparse for new scans in open SPECFile instance
2
3# reread data method (1)
4s.scan10.ReadData()
5scan10data = s.scan10.data
6
7# reread data method (2)
8s.Save2HDF5(h5) # save content of SPEC file to HDF5 file
9# read data from HDF5 file
10[angle1, angle2], scan10data = xu.io.geth5_scan(h5file, [10],
11 "motorname1",
12 "motorname2")
Reading EDF files
EDF files are mostly used to store CCD frames at ESRF recorded from various different detectors. This format is therefore used in combination with SPEC files. In an example the EDFFile class is used to parse the data from EDF files and store them to an HDF5 file. HDF5 if perfectly suited because it can handle large amount of data and compression.
1import xrayutilities as xu
2import numpy
3
4specfile = "specfile.spec"
5h5file = "h5file.h5"
6
7s = xu.io.SPECFile(specfile)
8s.Save2HDF5(h5file) # save to hdf5 file
9
10# read ccd frames from EDF files
11for i in range(1, 1001, 1):
12 efile = "edfdir/sample_%04d.edf" % i
13 e = xu.io.edf.EDFFile(efile)
14 e.ReadData()
15 e.Save2HDF5(h5file, group="/frelon_%04d" % i)
See also
the fully working example provided in the examples directory perfectly suited for reading data from beamline ID01
Reading XRDML files
Files recorded by Panalytical diffractometers in the .xrdml format can be parsed.
All supported file formats can also be parsed transparently when they are saved as compressed files using common compression formats. The parsing of such compressed .xrdml files conversion to reciprocal space and visualization by gridding is shown below:
1import xrayutilities as xu
2om, tt, psd = xu.io.getxrdml_map('rsm_%d.xrdml.bz2', [1, 2, 3, 4, 5],
3 path='data')
4# or using the more flexible function
5tt, om, psd = xu.io.getxrdml_scan('rsm_%d.xrdml.bz2', 'Omega',
6 scannrs=[1, 2, 3, 4, 5], path='data')
7# single files can also be opened directly using the low level approach
8xf = xu.io.XRDMLFile('data/rsm_1.xrdml.bz2')
9# then see xf.scan and its attributes
See also
the fully working example provided in the examples directory
Other formats
Other formats which can be read include
Rigaku
.rasfiles.files produces by the experimental control software at Hasylab/Desy (spectra).
numor files from the ILL neutron facility
ccd images in the tiff file format produced by RoperScientific CCD cameras and Perkin Elmer detectors.
files from recorded by Seifert diffractometer control software (
.nja)support is also provided for reading of
ciffiles from structure databases to extract unit cell parameters as well es read data from those files (pdCIF, ESG files)
See the examples directory for more information and working example scripts.
Angle calculation using Experiment and materials classes
Methods for high angle x-ray diffraction experiments. Mostly for experiments performed in coplanar scattering geometry. An example will be given for the calculation of the position of Bragg reflections.
1>>> import xrayutilities as xu
2>>> Si = xu.materials.Si # load material from materials submodule
3>>>
4>>> # initialize experimental class with directions from experiment
5>>> hxrd = xu.HXRD(Si.Q(1, 1, -2), Si.Q(1, 1, 1))
6>>> # calculate angles of Bragg reflections and print them to the screen
7>>> om, chi, phi, tt = hxrd.Q2Ang(Si.Q(1, 1, 1))
8>>> print("Si (111)")
9Si (111)
10>>> print(f"om, tt: {om:8.3f} {tt:8.3f}")
11om, tt: 14.221 28.442
12>>> om, chi, phi, tt = hxrd.Q2Ang(Si.Q(2, 2, 4))
13>>> print("Si (224)")
14Si (224)
15>>> print(f"om, tt: {om:8.3f} {tt:8.3f}")
16om, tt: 63.485 88.028
Note that above the HXRD class is initialized without specifying the energy used in the experiment. It will use the default energy stored in the configuration file, which defaults to CuK-\({\alpha}_1\).
One could also call:
hxrd = xu.HXRD(Si.Q(1, 1, -2), Si.Q(1, 1, 1), en=10000) # energy in eV
to specify the energy explicitly.
The HXRD class by default describes a four-circle goniometer as described in more detail here.
Similar functions exist for other experimental geometries. For grazing incidence diffraction one might use
1>>> import xrayutilities as xu
2>>> gid = xu.GID(xu.materials.Si.Q(1, -1, 0), xu.materials.Si.Q(0, 0, 1))
3>>> # calculate angles and print them to the screen
4>>> (alphai, azimuth, tt, beta) = gid.Q2Ang(xu.materials.Si.Q(2, -2, 0))
5>>> print(f"azimuth, tt: {azimuth:8.3f} {tt:8.3f}")
6azimuth, tt: 113.651 47.302
There is on implementation of a GID 2S+2D diffractometer. Be sure to check if the order of the detector circles fits your goniometer, otherwise define one yourself!
There exists also a powder diffraction class, which is able to convert powder scans from angular to reciprocal space.
1import xrayutilities as xu
2import numpy
3energy = 'CuKa12'
4# creating powder experiment
5xup = xu.PowderExperiment(en=energy)
6theta = numpy.arange(0, 70, 0.01)
7q = xup.Ang2Q(theta)
More information about powdered materials can be obtained from the PowderDiffraction class. It contains information about peak positions and intensities
1>>> import xrayutilities as xu
2>>> print(xu.simpack.PowderDiffraction(xu.materials.In))
3Powder diffraction object
4-------------------------
5Powder-In (a: 3.2523, c: 4.9461, at0_In_2a_occupation: 1, at0_In_2a_biso: 0, volume: 1, )
6Lattice:
7139 tetragonal I4/mmm: a = 3.2523, b = 3.2523 c= 4.9461
8alpha = 90.000, beta = 90.000, gamma = 90.000
9Lattice base:
100: In (49) 2a occ=1.000 b=0.000
11Reflection conditions:
12 general: hkl: h+k+l=2n, hk0: h+k=2n, 0kl: k+l=2n, hhl: l=2n, 00l: l=2n, h00: h=2n
132a : None
14
15Reflections:
16------------
17 h k l | tth | |Q| |Int | Int (%)
18 ---------------------------------------------------------------
19 (1, 0, 1) 32.9339 2.312 217.24 100.00
20 (0, 0, 2) 36.2964 2.541 41.69 19.19
21 (1, 1, 0) 39.1392 2.732 67.54 31.09
22 (1, 1, -2) 54.4383 3.731 50.58 23.28
23 (2, 0, 0) 56.5486 3.864 22.47 10.34
24 (1, 0, -3) 63.1775 4.273 31.82 14.65
25 (2, 1, 1) 67.0127 4.503 53.09 24.44
26 (2, 0, 2) 69.0720 4.624 24.22 11.15
27 (0, 0, 4) 77.0641 5.081 4.43 2.04
28 (2, 2, 0) 84.1193 5.464 7.13 3.28
29 (2, 1, -3) 89.8592 5.761 24.97 11.49
30 (1, 1, 4) 90.0301 5.769 12.44 5.73
31 (3, 0, 1) 93.3390 5.933 11.75 5.41
32 (2, -2, 2) 95.2543 6.026 11.43 5.26
33 (3, 1, 0) 97.0033 6.109 11.19 5.15
34 (2, 0, -4) 102.9976 6.384 10.69 4.92
35 (3, 1, 2) 108.4189 6.617 21.19 9.75
36 (1, 0, 5) 108.9602 6.639 10.60 4.88
37 (3, 0, 3) 116.5074 6.936 11.01 5.07
38 (2, -3, 1) 120.4651 7.081 22.87 10.53
39 (2, 2, -4) 132.3519 7.462 13.70 6.31
40 (0, 0, 6) 138.2722 7.622 3.88 1.79
41 (2, 1, -5) 140.6857 7.681 32.94 15.16
42 (4, 0, 0) 142.6631 7.728 8.67 3.99
43 (3, 2, -3) 153.5192 7.940 48.97 22.54
44 (3, 1, 4) 153.9051 7.946 49.71 22.88
45 (4, 1, 1) 162.8984 8.066 76.17 35.07
46 (1, 1, 6) 166.0961 8.097 46.91 21.60
47 (4, 0, 2) 171.5398 8.135 77.25 35.56
If you are interested in simulations of powder diffraction patterns look at section Powder diffraction simulations
Using the Gridder classes
xrayutilities provides Gridder classes for 1D, 2D, and 3D data sets. These Gridders map irregular spaced data onto a regular grid. This is often needed after transforming data measured at equally spaced angular positions to reciprocal space were their spacing is irregular.
In 1D this process actually equals the calculation of a histogram. Below you find the most basic way of using the Gridder in 2D. Other dimensions work very similar.
The most easiest use (what most user might need) is
1import xrayutilities as xu # import Python package
2g = xu.Gridder2D(100, 101) # initialize the Gridder object, which will
3# perform Gridding to a regular grid with 100x101 points
4#====== load some data here =====
5g(x, y, data) # call the gridder with the data
6griddata = g.data # the data attribute contains the gridded data.
A more complicated example showing also sequential gridding is shown below. You need sequential gridding when you can not load all data at the same time, which is often problematic with 3D data sets. In such cases you need to specify the data range before the first call to the gridder.
1import xrayutilities as xu # import Python package
2g = xu.Gridder2D(100, 101) # initialize the Gridder object
3g.KeepData(True)
4g.dataRange(1, 2, 3, 4) # (xgrd_min, xgrd_max, ygrd_min, ygrd_max)
5#====== load some data here =====
6g(x, y, data) # call the gridder with the data
7griddata = g.data # the data attribute contains the so far gridded data.
8
9#====== load some more data here =====
10g(x, y, data) # call the gridder with the new data
11griddata = g.data # the data attribute contains the combined gridded data.
Gridder2D for visualization
Based on the example of parsed data from XRDML files shown above (Reading XRDML files) we show here how to use the Gridder2D class together with matplotlibs contourf.
1Si = xu.materials.Si
2hxrd = xu.HXRD(Si.Q(1, 1, 0), Si.Q(0, 0, 1))
3qx, qy, qz = hxrd.Ang2Q(om, tt)
4gridder = xu.Gridder2D(200, 600)
5gridder(qy, qz, psd)
6INT = xu.maplog(gridder.data.transpose(), 6, 0)
7# plot the intensity as contour plot
8plt.figure()
9cf = plt.contourf(gridder.xaxis, gridder.yaxis, INT, 100, extend='min')
10plt.xlabel(r'$Q_{[110]}$ ($\mathrm{\AA^{-1}}$)')
11plt.ylabel(r'$Q_{[001]}$ ($\mathrm{\AA^{-1}}$)')
12cb = plt.colorbar(cf)
13cb.set_label(r"$\log($Int$)$ (cps)")
14plt.tight_layout()
The shown script results in the plot of the reciprocal space map shown below.
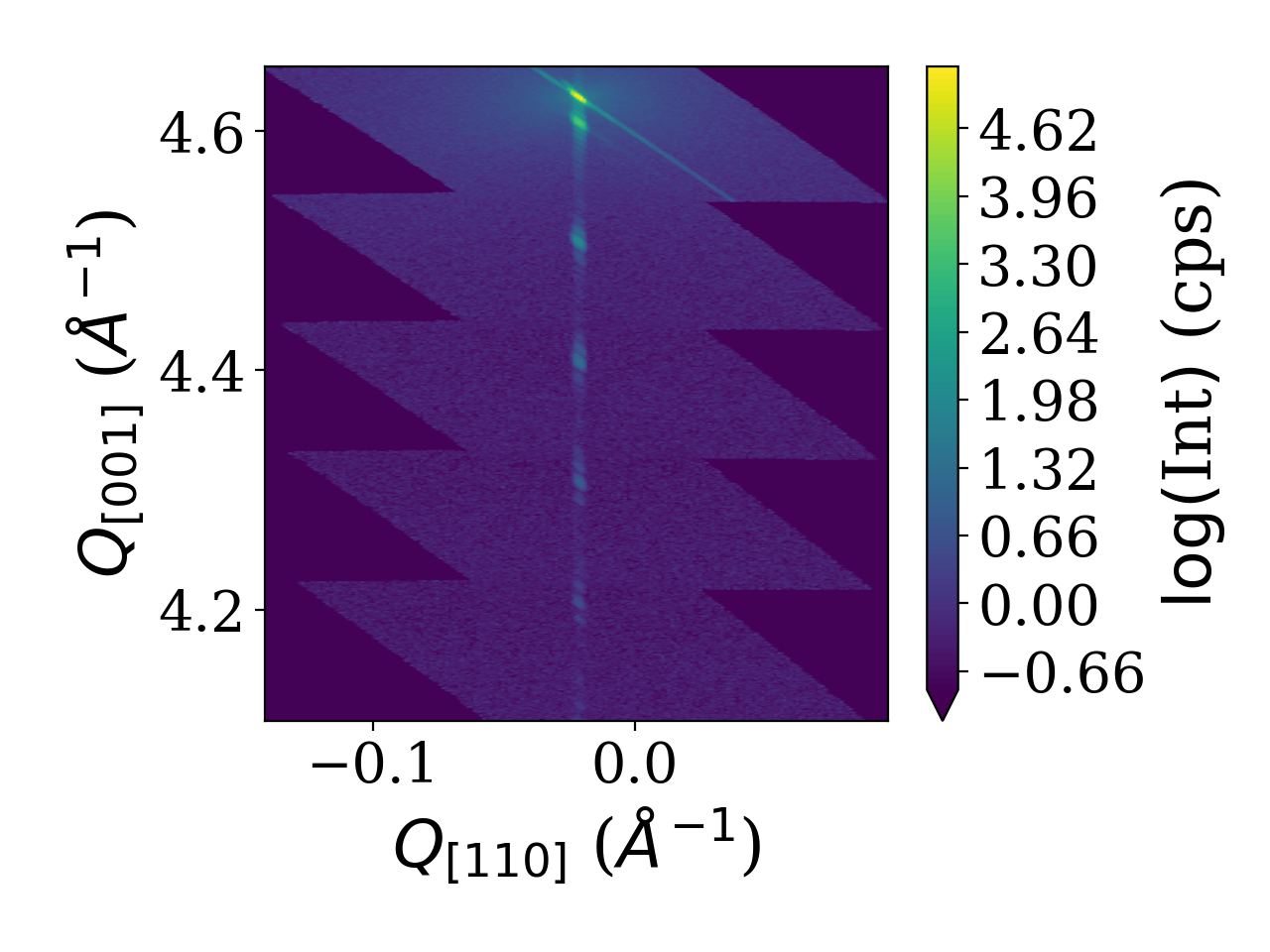
Line cuts from reciprocal space maps
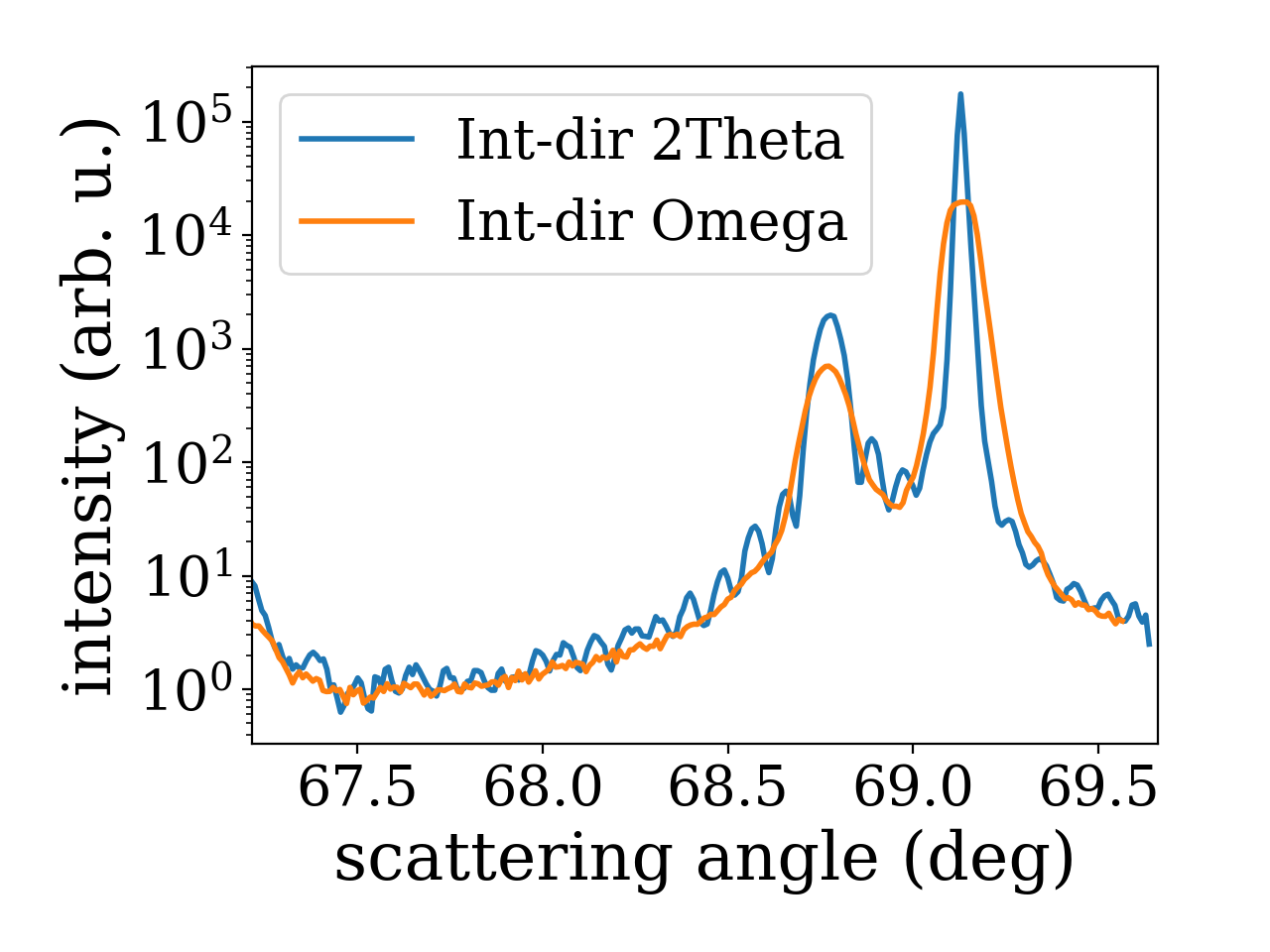
Using the analysis subpackage one can produce line cuts. Starting from the reciprocal space data produced by the reciprocal space conversion as in the last example code we extract radial scan along the crystal truncation rod. For the extraction of line scans the respective functions offer to integrate the data along certain directions. In the present case integration along ‘2Theta’ gives the best result since a broadening in that direction was caused by the beam footprint in the particular experiment. For different line cut functions various integration directions are possible. They are visualized in the figure below.
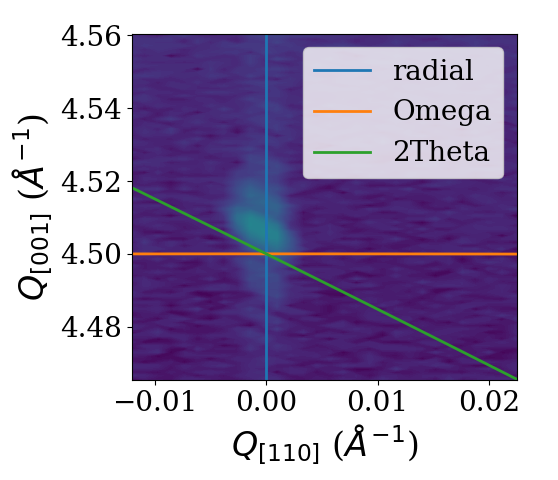
1# line cut with integration along 2theta to remove beam footprint broadening
2qzc, qzint, cmask = xu.analysis.get_radial_scan([qy, qz], psd, [0, 4.5],
3 1001, 0.155, intdir='2theta')
4
5# line cut with integration along omega
6qzc_om, qzint_om, cmask_om = xu.analysis.get_radial_scan([qy, qz], psd, [0, 4.5],
7 1001, 0.155, intdir='omega')
8plt.figure()
9plt.semilogy(qzc, qzint, label='Int-dir 2Theta')
10plt.semilogy(qzc_om, qzint_om, label='Int-dir Omega')
11plt.xlabel(r'scattering angle (deg)')
12plt.ylabel(r'intensity (arb. u.)')
13plt.legend()
14plt.tight_layout()
Using the materials subpackage
xrayutilities provides a set of Python classes to describe crystal lattices and materials.
Examples show how to define a new material by defining its lattice and deriving a new material, furthermore materials can be used to calculate the structure factor of a Bragg reflection for an specific energy or the energy dependency of its structure factor for anomalous scattering. Data for this are taken from a database which is included in the download.
First defining a new material from scratch is shown. This is done from the space group and Wyckhoff positions of the atoms inside the unit cell. Depending on the space group number the initialization of a new SGLattice object expects a different amount of parameters. For a cubic materials only the lattice parameter a should be given while for a triclinic materials a, b, c, alpha, beta, and gamma have to be specified. Its similar for the Wyckoff positions. While some Wyckoff positions require only the type of atom others have some free paramters which can be specified. Below we show the definition of zincblende InP as well as for its hexagonal wurtzite polytype together with a quick visualization of the unit cells. A more accurate visualization of the unit cell can be performed when using show_unitcell() with the Mayavi mode or by using the CIF-exporter and an external tool.
1import matplotlib.pyplot as plt
2import xrayutilities as xu
3
4# elements (which contain their x-ray optical properties) are loaded from
5# xrayutilities.materials.elements
6In = xu.materials.elements.In
7P = xu.materials.elements.P
8
9# define elastic parameters of the material we use a helper function which
10# creates the 6x6 tensor needed from the only 3 free parameters of a cubic
11# material.
12elastictensor = xu.materials.CubicElasticTensor(10.11e+10, 5.61e+10,
13 4.56e+10)
14# definition of zincblende InP:
15InP = xu.materials.Crystal(
16 "InP", xu.materials.SGLattice(216, 5.8687, atoms=[In, P],
17 pos=['4a', '4c']),
18 elastictensor)
19
20# a hexagonal equivalent which shows how parameters change for material
21# definition with a different space group. Since the elasticity tensor is
22# optional its not specified here.
23InPWZ = xu.materials.Crystal(
24 "InP(WZ)", xu.materials.SGLattice(186, 4.1423, 6.8013,
25 atoms=[In, P], pos=[('2b', 0),
26 ('2b', 3/8.)]))
27f = plt.figure()
28InP.show_unitcell(fig=f, subplot=121)
29plt.title('InP zincblende')
30InPWZ.show_unitcell(fig=f, subplot=122)
31plt.title('InP wurtzite')
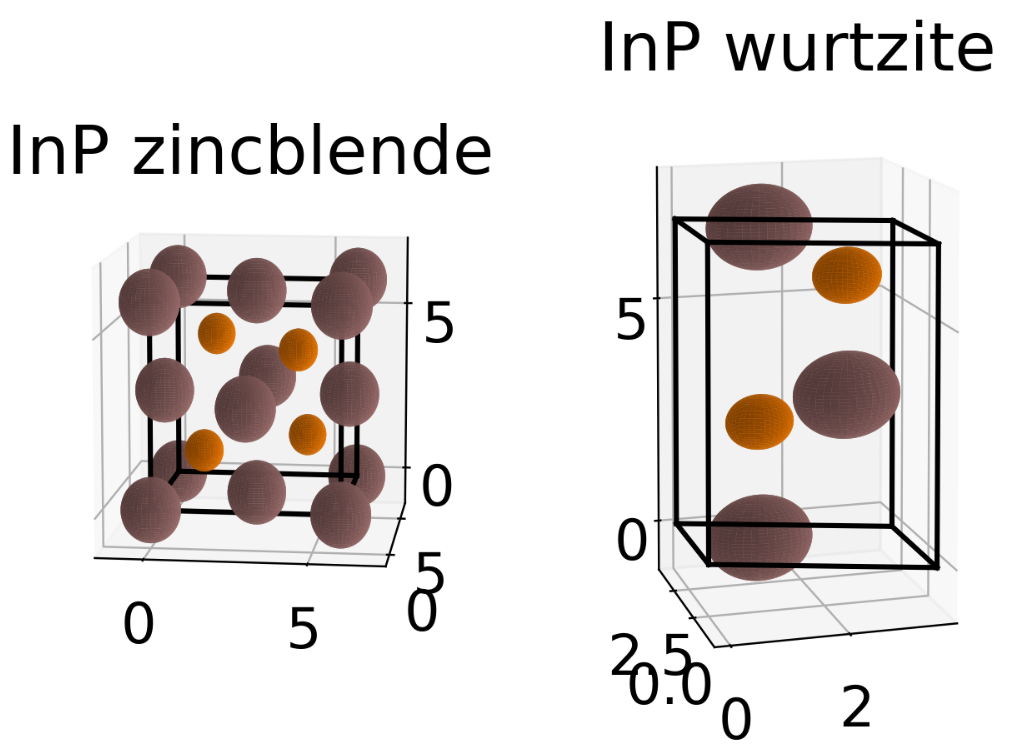
InP (in both variants) is already included in the xu.materials module and can be loaded by
InP = xu.materials.InP
InPWZ = xu.materials.InPWZ
Similar definitions exist for many other materials. Alternatively to giving the Wyckoff labels and parameters one can also specify the position of one atom for every unique site within the unit cell. xrayutilities will then search the corresponding Wyckoff position of this atom and populate therefore populate all equivalent sites as well. For the example of InP in zincblende form the material definition could also look as shown below. Note that instead of the elements also the elemental symbol as string can be used:
InP = xu.materials.Crystal(
"InP", xu.materials.SGLattice(216, 5.8687, atoms=["In", "P"],
pos=[(0,0,0), (1/4, 1/4, 1/4)]))
Using the material properties the calculation of the reflection strength of a Bragg reflection can be done as follows
1import xrayutilities as xu
2
3# defining material and experimental setup
4InAs = xu.materials.InAs
5energy= 8048 # eV
6
7# calculate the structure factor for InAs (111) (222) (333)
8hkllist = [[1, 1, 1], [2, 2, 2], [3, 3, 3]]
9for hkl in hkllist:
10 qvec = InAs.Q(hkl)
11 F = InAs.StructureFactor(qvec, energy)
12 print(f"|F| = {abs(F):8.3f}")
Similar also the energy dependence of the structure factor can be determined
1import matplotlib.pyplot as plt
2
3energy= numpy.linspace(500, 20000, 5000) # 500 - 20000 eV
4F = InAs.StructureFactorForEnergy(InAs.Q(1, 1, 1), energy)
5
6plt.figure(); plt.clf()
7plt.plot(energy, F.real, '-k', label='Re(F)')
8plt.plot(energy, F.imag, '-r', label='Imag(F)')
9plt.xlabel("Energy (eV)"); plt.ylabel("F"); plt.legend()
It is also possible to calculate the components of the structure factor of atoms, which may be needed for input into XRD simulations.
1# f = f0(|Q|) + f1(en) + j * f2(en)
2import xrayutilities as xu
3
4Fe = xu.materials.elements.Fe # iron atom
5Q = [0, 0, 1.9]
6en = 10000 # energy in eV
7
8print(f"Iron (Fe): E: {en:9.1f} eV")
9print(f"f0: {Fe.f0(xu.math.VecNorm(Q)):8.4g}")
10print(f"f1: {Fe.f1(en):8.4g}")
11print(f"f2: {Fe.f2(en):8.4g}")
Iron (Fe): E: 10000.0 eV
f0: 21.78
f1: -0.0178
f2: 2.239
Transformation of SGLattice
SGLattice-objects can be transformed to use a different unit cell setting. This can be used to for example change the origin choice after the material definition or to convert into a totally different setting, e.g. for simulation purposes.
The code below shows the example of the Diamond structure converted between the two different origin choices
1import numpy
2import xrayutilities as xu
3C1 = xu.materials.SGLattice("227:1", 3.5668, atoms=["C"],
4 pos=[(0,0,0)])
5C2 = xu.materials.SGLattice("227:2", 3.5668, atoms=["C"],
6 pos=[(1/8, 1/8, 1/8)])
7C1 == C2 # False
8C3 = C2.transform(numpy.identity(3), (1/8, 1/8, 1/8))
9C3 == C1 # True
For dynamical diffraction simulations of cubic crystals with (111) surface it might be required to convert the unit cell in a way that a principle axis is pointing along the surface normal. Using an apropriate conversion matrix this is shown for the example of InP
1import xrayutilities as xu
2InP111_lattice = xu.materials.InP.lattice.transform(((-1/2, 0, 1),
3 (1/2, -1/2, 1),
4 (0, 1/2, 1)),
5 (0, 0, 0))
While the built in InP uses the cubic setting with space group F-43m(#216) the converted lattice has rhombohedral space group (in this case R3m(#160)) and converted atomic positions.
Visualization of the Bragg peaks in a reciprocal space plane
If you want to explore which peaks are available and reachable in coplanar diffraction geometry and what their relationship between different materials is xrayutilities provides a function which generates a slightly interactive plot which helps you with this task.
1import xrayutilities as xu
2mat = xu.materials.Crystal('GaTe',
3 xu.materials.SGLattice(194, 4.06, 16.96,
4 atoms=['Ga', 'Te'],
5 pos=[('4f', 0.17),
6 ('4f', 0.602)]))
7ttmax = 160
8sub = xu.materials.Si
9hsub = xu.HXRD(sub.Q(1, 1, -2), sub.Q(1, 1, 1))
10ax, h = xu.materials.show_reciprocal_space_plane(sub, hsub, ttmax=160)
11hxrd = xu.HXRD(mat.Q(1, 0, 0), mat.Q(0, 0, 1))
12ax, h2 = xu.materials.show_reciprocal_space_plane(mat, hxrd, ax=ax)
The generated plot shows all the existing Bragg spots, their (hkl) label is shown when the mouse is over a certain spot and the diffraction angles calculated by the given HXRD object is printed when you click on a certain spot. Not that the primary beam is assumed to come from the left, meaning that high incidence geometry occurs for all peaks with positive inplane momentum transfer.
![cut of reciprocal space for cubic Si(111) and a hexagonal material with c-axis along [111] of the substrate](_images/reciprocal_space_plane.png)
Calculation of diffraction angles for a general geometry
Often the restricted predefined geometries are not corresponding to the experimental setup, nevertheless xrayutilities is able to calculate the goniometer angles needed to reach a certain reciprocal space position.
For this purpose the goniometer together with the geometric restrictions need to be defined and the q-vector in laboratory reference frame needs to be specified. This works for arbitrary goniometer, however, the user is expected to set up bounds to put restrictions to the number of free angles to obtain reproducible results. In general only three angles are needed to fit an arbitrary q-vector (2 sample + 1 detector angles or 1 sample + 2 detector). More goniometer angles can be kept free if some pseudo-angle constraints are used instead.
The example below shows the necessary code to perform such an angle calculation for a custom defined material with orthorhombic unit cell.
1import xrayutilities as xu
2import numpy as np
3
4def Pnma(a, b, c):
5 # create orthorhombic unit cell with space-group 62,
6 # here for simplicity without base
7 l = xu.materials.SGLattice(62, a, b, c)
8 return l
9
10latticeConstants=[5.600, 7.706, 5.3995]
11SmFeO3 = xu.materials.Crystal("SmFeO3", Pnma(*latticeConstants))
12# 2S+2D goniometer
13qconv=xu.QConversion(('x+', 'z+'), ('z+', 'x+'), (0, 1, 0))
14# [1,1,0] surface normal
15hxrd = xu.Experiment(SmFeO3.Q(0, 0, 1), SmFeO3.Q(1, 1, 0), qconv=qconv)
16
17hkl=(2, 0, 0)
18q_material = SmFeO3.Q(hkl)
19q_laboratory = hxrd.Transform(q_material) # transform
20
21print(f"SmFeO3: hkl {hkl}, qvec {np.round(q_material, 5)}")
22print(f"Lattice plane distance: {SmFeO3.planeDistance(hkl):.4f}")
23
24#### determine the goniometer angles with the correct geometry restrictions
25# tell bounds of angles / (min,max) pair or fixed value for all motors.
26# maximum of three free motors! here the first goniometer angle is fixed.
27# om, phi, tt, delta
28bounds = (5, (-180, 180), (-1, 90), (-1, 90))
29ang, qerror, errcode = xu.Q2AngFit(q_laboratory, hxrd, bounds)
30print(f"err {errcode} ({qerror:.3g}) angles {np.round(ang, 5)}")
31# check that qerror is small!!
32print("sanity check with back-transformation (hkl): ",
33 np.round(hxrd.Ang2HKL(*ang,mat=SmFeO3),5))
The output of the code above would be similar to:
SmFeO3: hkl (2, 0, 0), qvec [2.24399 0. 0. ]
Lattice plane distance: 2.8000
err 0 (9.61e-09) angles [ 5. 20.44854 19.65315 25.69328]
sanity check with back-transformation (hkl): [ 2. 0. -0.]
In the example above all angles can be kept free if a pseudo-angle constraint is used in addition.
This is shown below for the incidence angle, which when fixed to 5 degree results in the same goniometer angles as shown above.
Currently two helper functions for incidence and exit angles (incidenceAngleConst() and exitAngleConst()) are implemented, but user-defined functions can be supplied.
1aiconstraint = xu.q2ang_fit.incidenceAngleConst
2bounds = ((0, 90), (-180, 180), (-1, 90), (-1, 90))
3ang, qerror, errcode = xu.Q2AngFit(
4 q_laboratory, hxrd, bounds,
5 constraints=[{'type':'eq', 'fun': lambda a: aiconstraint(a, 5, hxrd)}, ])
User-specific config file
Several options of xrayutilities can be changed by options in a config file. This includes the default x-ray energy as well as parameters to set the number of threads used by the parallel code and the verbosity of the output.
The default options are stored inside the installad Python module and should not be changed. Instead it is suggested to use a user-specific config file ‘~/.xrayutilities.conf’ or a ‘xrayutilities.conf’ file in the working directory.
An example of such a user config file is shown below:
1# begin of xrayutilities configuration
2[xrayutilities]
3
4# verbosity level of information and debugging outputs
5# 0: no output
6# 1: very import notes for users
7# 2: less import notes for users (e.g. intermediate results)
8# 3: debuging output (e.g. print everything, which could be interesing)
9# levels can be changed in the config file as well
10verbosity = 1
11
12# default wavelength in angstrom,
13wavelength = MoKa1 # Molybdenum K alpha1 radiation (17479.374eV)
14
15# default energy in eV
16# if energy is given wavelength settings will be ignored
17#energy = 10000 #eV
18
19# number of threads to use in parallel sections of the code
20nthreads = 1
21# 0: the maximum number of available threads will be used (as returned by
22# omp_get_max_threads())
23# n: n-threads will be used
Determining detector parameters
In the following three examples of how to determine the detector parameters for linear and area detectors is given. The procedure we use is in more detail described in this article.
Linear detectors
To determine the detector parameters of a linear detector one needs to perform a scan with the detector angle through the primary beam and aquire a detector spectrum at any point.
Using the following script determines the parameters necessary for the detector initialization, which are:
pixelwidth of one channel
the center channel
and the detector tilt (optional)
1"""
2example script to show how the detector parameters
3such as pixel width, center channel and detector tilt
4can be determined for a linear detector.
5"""
6
7import os
8
9import xrayutilities as xu
10
11# load any data file with with the detector spectra of a reference scan
12# in the primary beam, here I use spectra measured with a Seifert XRD
13# diffractometer
14dfile = os.path.join("data", "primarybeam_alignment20130403_2_dis350.nja")
15s = xu.io.SeifertScan(dfile)
16
17ang = s.axispos["T"] # detector angles during the scan
18spectra = s.data[:, :, 1] # detector spectra aquired
19
20# determine detector parameters
21# this function accepts some optional arguments to describe the goniometer
22# see the API documentation
23pwidth, cch, tilt = xu.analysis.linear_detector_calib(ang, spectra,
24 usetilt=True)
Area detector (Variant 1)
To determine the detector parameters of a area detector one needs to perform scans with the detector angles through the primary beam and aquire a detector images at any position. For the area detector at least two scans (one with the outer detector and and one with the inner detector angle) are required.
Using the following script determines the parameters necessary for the detector initialization from such scans in the primary beam only. Further down we discuss an other variant which is also able to use additionally detector images recorded at the Bragg reflection of a known reference crystal.
The determined detector parameters are:
center channels: position of the primary beam at the true zero position of the goniometer (considering the outer angle offset) (2 parameters)
pixelwidth of the channels in both directions (2 parameters), these two parameters can be replaced by the detector distance (1 parameter) if the pixel size is given as an input
detector tilt azimuth in degree from 0 to 360
detector tilt angle in degree (>0deg)
detector rotation around the primary beam in degree
outer angle offset, which describes a offset of the outer detector angle from its true zero position
The misalignment parameters as well as the pixel size can be fixed during the fitting.
1"""
2example script to show the detector parameter determination for area detectors
3from images recorded in the primary beam
4"""
5
6import os
7
8import xrayutilities as xu
9
10en = 10300.0 # eV
11datadir = os.path.join("data", "wire_") # data path for CCD files
12# template for the CCD file names
13filetmp = os.path.join(datadir, "wire_12_%05d.edf.gz")
14
15# manually selected images
16# select images which have the primary beam fully on the CCD
17imagenrs = [2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19,
18 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33]
19
20images = []
21ang1 = []
22ang2 = []
23
24# read images and angular positions from the data file
25# this might differ for data taken at different beamlines since
26# they way how motor positions are stored is not always consistent
27for imgnr in imagenrs:
28 filename = filetmp % imgnr
29 edf = xu.io.EDFFile(filename)
30 images.append(edf.data)
31 ang1.append(float(edf.header['ESRF_ID01_PSIC_NANO_NU']))
32 ang2.append(float(edf.header['ESRF_ID01_PSIC_NANO_DEL']))
33
34
35# call the fit for the detector parameters
36# detector arm rotations and primary beam direction need to be given.
37# in total 9 parameters are fitted, however the severl of them can
38# be fixed. These are the detector tilt azimuth, the detector tilt angle, the
39# detector rotation around the primary beam and the outer angle offset
40# The detector pixel size or the detector distance should be kept unfixed to
41# be optimized by the fit.
42param, eps = xu.analysis.sample_align.area_detector_calib(
43 ang1, ang2, images, ['z+', 'y-'], 'x+',
44 start=(None, None, 1.0, 45, 0, -0.7, 0),
45 fix=(False, False, True, False, False, False, False),
46 wl=xu.en2lam(en))
A possible output of this script could be
fitted parameters: epsilon: 8.0712e-08 (2,[‘Parameter convergence’]) param: (cch1,cch2,pwidth1,pwidth2,tiltazimuth,tilt,detrot,outerangle_offset) param: 140.07 998.34 4.4545e-05 4.4996e-05 72.0 1.97 -0.792 -1.543 please check the resulting data (consider setting plot=True) detector rotation axis / primary beam direction (given by user): [‘z+’, ‘y-’] / x+ detector pixel directions / distance: z- y+ / 1 detector initialization with: init_area(‘z-’, ‘y+’, cch1=140.07, cch2=998.34, Nch1=516, Nch2=516, pwidth1=4.4545e-05, pwidth2=4.4996e-05, distance=1., detrot=-0.792, tiltazimuth=72.0, tilt=1.543) AND ALWAYS USE an (additional) OFFSET of -1.9741deg in the OUTER DETECTOR ANGLE!
The output gives the fitted detector parameters and compiles the Python code line one needs to use to initialize the detector. Important to note is that the outer angle offset which was determined by the fit (-1.9741 degree in the aboves example) is not included in the initialization of the detector parameters but needs to be used in every call to the q-conversion function as offset. This step needs to be performed manually by the user!
Area detector (Variant 2)
In addition to scans in the primary beam this variant enables also the use of detector images recorded in scans at Bragg reflections of a known reference materials. However this also required that the sample orientation and x-ray wavelength need to be fit. To keep the additional parameters as small as possible we only implemented this for symmetric coplanar diffractions.
The advantage of this method is that it is more sensitive to the outer angle offset also at large detector distances. The additional parameters are:
sample tilt angle in degree
sample tilt azimuth in degree
and the x-ray wavelength in angstrom
1"""
2example script to show the detector parameter determination for area detectors
3from images recorded in the primary beam and at known symmetric coplanar Bragg
4reflections of a reference crystal
5"""
6
7import os
8
9import numpy
10import xrayutilities as xu
11
12Si = xu.materials.Si
13
14datadir = 'data'
15specfile = "si_align.spec"
16
17en = 15000 # eV
18wl = xu.en2lam(en)
19imgdir = os.path.join(datadir, "si_align_") # data path for CCD files
20filetmp = "si_align_12_%04d.edf.gz"
21
22qconv = xu.QConversion(['z+', 'y-'], ['z+', 'y-'], [1, 0, 0])
23hxrd = xu.HXRD(Si.Q(1, 1, -2), Si.Q(1, 1, 1), wl=wl, qconv=qconv)
24
25# manually selected images
26
27s = xu.io.SPECFile(specfile, path=datadir)
28imagenrs = []
29for num in [61, 62, 63, 20, 21, 26, 27, 28]:
30 s[num].ReadData()
31 imagenrs = numpy.append(imagenrs, s[num].data['ccd_n'])
32
33# avoid images which do not have to full beam on the detector as well as
34# other which show signal due to cosmic radiation
35avoid_images = [37, 57, 62, 63, 65, 87, 99, 106, 110, 111, 126, 130, 175,
36 181, 183, 185, 204, 206, 207, 208, 211, 212, 233, 237, 261,
37 275, 290]
38
39images = []
40ang1 = [] # outer detector angle
41ang2 = [] # inner detector angle
42sang = [] # sample rocking angle
43hkls = [] # Miller indices of the reference reflections
44
45
46def hotpixelkill(ccd):
47 """
48 function to remove hot pixels from CCD frames
49 ADD REMOVE VALUES IF NEEDED!
50 """
51 ccd[304, 97] = 0
52 ccd[303, 96] = 0
53 return ccd
54
55
56# read images and angular positions from the data file
57# this might differ for data taken at different beamlines since
58# they way how motor positions are stored is not always consistent
59for imgnr in numpy.sort(list(set(imagenrs) - set(avoid_images))[::4]):
60 filename = os.path.join(imgdir, filetmp % imgnr)
61 edf = xu.io.EDFFile(filename)
62 ccd = hotpixelkill(edf.data)
63 images.append(ccd)
64 ang1.append(float(edf.header['motor_pos'].split()[4]))
65 ang2.append(float(edf.header['motor_pos'].split()[3]))
66 sang.append(float(edf.header['motor_pos'].split()[1]))
67 if imgnr > 1293.:
68 hkls.append((0, 0, 0))
69 elif imgnr < 139:
70 hkls.append((0, 0, numpy.sqrt(27))) # (3,3,3))
71 else:
72 hkls.append((0, 0, numpy.sqrt(75))) # (5,5,5))
73
74# call the fit for the detector parameters.
75# Detector arm rotations and primary beam direction need to be given
76# in total 8 detector parameters + 2 additional parameters for the reference
77# crystal orientation and the wavelength are fitted, however the 4 misalignment
78# parameters of the detector and the 3 other parameters can be fixed.
79# The fixable parameters are detector tilt azimuth, the detector tilt angle,
80# the detector rotation around the primary beam, the outer angle offset, sample
81# tilt, sample tilt azimuth and the x-ray wavelength
82# Additionally if accurately known the detector pixel size can be given and
83# fixed and instead the detector distance can be fitted.
84param, eps = xu.analysis.area_detector_calib_hkl(
85 sang, ang1, ang2, images, hkls, hxrd, Si, ['z+', 'y-'], 'x+',
86 start=(None, None, 1.0, 45, 1.69, -0.55, -1.0, 1.3, 60., wl),
87 fix=(False, False, True, False, False, False, False, False, False, False),
88 plot=True)
89
90# Following is an example of the output of the summary of the
91# area_detector_calib_hkl function
92# total time needed for fit: 624.51sec
93# fitted parameters: epsilon: 9.9159e-08 (2,['Parameter convergence'])
94# param:
95# (cch1,cch2,pwidth1,pwidth2,tiltazimuth,tilt,detrot,outerangle_offset,
96# sampletilt,stazimuth,wavelength)
97# param: 367.12 349.27 6.8187e-05 6.8405e-05 131.4 2.87 -0.390 -0.061 1.201
98# 318.44 0.8254
99# please check the resulting data (consider setting plot=True)
100# detector rotation axis / primary beam direction (given by user): ['z+', 'y-']
101# / x+
102# detector pixel directions / distance: z- y+ / 1
103# detector initialization with:
104# init_area('z-', 'y+', cch1=367.12, cch2=349.27, Nch1=516, Nch2=516,
105# pwidth1=6.8187e-05, pwidth2=6.8405e-05, distance=1., detrot=-0.390,
106# tiltazimuth=131.4, tilt=2.867)
107# AND ALWAYS USE an (additional) OFFSET of -0.0611deg in the OUTER
108# DETECTOR ANGLE!